
Translate this page into:
Modulation of the Extracellular Signal-Regulated Protein Kinase and Tissue Inhibitors of Matrix Metalloproteases-1 Gene in Chronic Neuropathic Pain

-
Received: ,
Accepted: ,
How to cite this article: Saxena AK, Khrolia D, Chilkoti GT, Gondode PG, Sharma T, Thakur G, et al. Modulation of the extracellular signal-regulated protein kinase and tissue inhibitors of matrix metalloproteases-1 gene in chronic neuropathic pain. Indian J Palliat Care 2021;27(2):251-6.
Abstract
Objectives:
The aim of this study is to study the modulation of extracellular signal-regulated protein kinase (ERK) and tissue inhibitors of matrix metalloproteases 1 (TIMP 1) gene in patients with neuropathic pain (NP).
Materials and Methods:
In the present, cross-sectional, observational study, 2 ml of venous baseline sample was withdrawn from all the patients with neuropathic (NP) or non NP (NNP) soon after their diagnosis or on their first visit to the pain clinic. A real-time quantitative polymerase chain reaction experiment was conducted to measure the mRNA expression of TIMP1 and ERK genes in blood samples. The Delta Ct, Delta Ct, and fold change analysis of both the genes were conducted between patients with NP and NNP.
Results:
A total of 285 patients with chronic pain were assessed, out of which, 153 patients had NP and 132 had NNP. The average duration of chronic pain was 11 months for 285 patients. The mRNA expression of TIMP1 gene is significantly down regulated (2.65-fold) (P (-f. 01), and the mRNA expression level of ERK is significantly up regulated (2.03-fold) (P (-f. 01) in NP patients when compared with NNP.
Conclusion:
The mRNA expression of TIMP1 gene is significantly down regulated, and ERK is significantly up regulated in patients with NP. Further, multicentric trials with larger sample size are recommended to confirm this finding.
Keywords
Chronic neuropathic pain
Extracellular signal-regulated protein kinase
Tissue inhibitors of matrix metalloproteases 1

INTRODUCTION
Neuropathic pain (NP) arises as a consequence of a lesion or disease of the somatosensory nervous system and is a disabling clinical condition.[1,2] The prevalence of NP is estimated to be 3.3%–11.8% of the general population and is projected to increase with the aging population, diabetes epidemic, and improved cancer survival.[3-5] NP is also associated with a high level of disability and a large socioeconomic cost and chronic persistent pain being the most common complication.[6,7] The treatment options available is meager, and hence, emphasis has been laid upon the study of its molecular mechanism.[8]
A complete understanding of the various predictors influencing the occurrence and intensity of NeuP and the extent to which they depend on the specific underlying etiology is essential. Recently, genetic susceptibility has been known to contribute to the risk of developing NeuP. Evidence from human genetic studies have shown that NeuP disorders may be caused by mutations in a single gene or multiple genes thus leading to inter-individual variation in the susceptibility and intensity of NP.[9-12] A better understanding of the genetic architecture of NeuP will not only provide fundamental insight into the disease pathophysiology, but would also reveal new drug targets and thus help in the preemptive planning of the individual treatment plans.
Tissue inhibitors of matrix metalloproteases (TIMPs 1-4) are endogenously expressed proteins that can prevent the activation of all the pro forms of matrix metalloproteinase (MMPs). MMPs, a family of zinc-dependent extracellular proteases, have constitutive expression in uninjured states and mediates the cellular extracellular environment by assisting in growth and regenerative processes as it is expressed in migrating growth cones.[13-15] The balance between the expression of MMPs and their endogenous tissue inhibitors (TIMPs) has important implications in the pathogenesis and recovery in disease or peripheral nerve injury.[16] Various animals and human research have confirmed a lack of TIMP induction corresponding to MMP upregulation postinjury resulting in excess MMP proteolytic activity.[17]
Following nerve injury, phosphorylation of extracellular signal-regulated protein kinase (ERK), an important member of MAPK family, is sequentially increased in neurons, microglia, and astrocytes of the dorsal horn and gracile nucleus, and in injured large dorsal root ganglion neurons.[18] Nerve injury-induced phosphorylation of ERK occurs early and is long-lasting. In several animal models of neuropathic pain, mitogen-activated protein kinase (MAPK) inhibitors, known to suppress the synthesis of ERK, have proven effective to alleviate pain at various time points.
Thus, the modulation of TIMP and ERK/MAPK can be considered as a promising therapeutic target for the treatment of NP. On literature search, it was observed that the expression of TIMP1 gene has never been explored in human participants. However, the gene expression of ERK has been studied in a human study by Saxena et al. in patients with chronic persistent postsurgical pain, the mechanism of latter too is neuropathic in origin.[19] Therefore, the present study was undertaken to study the role of modulation of ERK and TIMP 1 gene in the pathogenesis of NP.
MATERIALS AND METHODS
This cross-sectional, observational study was undertaken following approval from the Institutional Ethics Committee for Human Research. The study was conducted in the Departments of Anesthesiology and Critical Care and Biochemistry. A written informed consent was taken from each patient included in the study. The present study was conducted between November 2016 and April 2018. Patients with established diagnosis of either NP (Neuropathic) or non NP (NNP), who were ≥18 years of age with pain duration of at least 3 months with pain intensity of ≥4 on 10 cm on Visual Analog Scale (VAS) were included. In all the patients, a baseline 2 ml venous sample was collected in an EDTA vial and stored at −20°C after fixing it in TRIZOL reagent for further gene expression studies.
The blood sample was labeled appropriately, stored under suitable conditions, and entered on the tissue register to ensure traceability. These standard operating procedures were in conjunction with the human tissue authority code of practice and standards.
Methodology for gene expression
For the estimation of the TIMP1 and ERK gene expression study, all the molecular biological studies were performed in a Laminar Air Cabinet (Toshiba, India). Estimation of TIMP1 and ERK gene expression in the blood was performed using a CFX connect Biorad two-color high-resolution melt (HRM) real-time polymerase chain reaction (RTPCR) in the following steps:
• Step 1: Extraction of RNA from blood sample:
RNA was isolated from whole blood using Trizol BD (Ambion, Invitrogen Bioservices, India) as per the manufacturer’s protocol. The concentration and purity of the samples were determined using a spectrophotometer (Nanodrop1000; Thermofisher, USA). All the samples had a ratio of absorbance at 260/280 between 1.8 and 2.0.
• Step 2: Synthesis of complementary DNA (cDNA):
From the total RNA extracted, mRNA was used as a template for the synthesis of cDNA. On the same day preferably, total RNA (1 μg) was converted into first strand cDNA using a Maxima first strand cDNA synthesis kit (Thermo USA) according to the manufacturer’s protocol.
• Step 3: Quantification of TIMP1 and ERK gene expression by two-color HRM RTPCR
A RT quantitative PCR (qPCR) experiment was conducted to measure the expression of TIMP1 and ERK genes in blood samples from the subjects. The qPCR reactions were performed on CFX connect Biorad two-color HRM RTPCR, following PCR conditions and primer sequences. Table 1 shows the primer sequences of genes used for the real-time PCR assay, whereas Table 2 shows the cycling conditions used for the RTPCR assay.
| Gene | Primer sequences |
|---|---|
| GAPDH | (Forward) 5’-AAGGGTCATCATCTCTGC-3’ (Reverse) 5’-TAAGCAGTTGGTGGTGCA-3’ |
| TIMP1 | (Forward) 5’CTGTTGTTGCTGTGGCTGATA -3’ (Reverse) 5’ACGCTGGTATAAGGTGGTCTG-3’ |
| ERK | (Forward) 5’-CCCGTCTTGGCTTATCCACT-3’ (Reverse) 5’-TACATACTGCCGCAGGTCAC-3’ |
ERK: Extracellular signal-regulated protein kinase
| Gene | Initial denaturation (min) | Denaturation (S) | Annealing/extension (S) | Cycles |
|---|---|---|---|---|
| GAPDH | 95°C/5 | 95°C/10 | 62.5°C/30 | 39 |
| TIMP1 | 95°C/5 | 95°C/10 | 54.0°C/30 | 39 |
| ERK | 95°C/5 | 95°C/10 | 62.0°C/20 | 39 |
ERK: Extracellular signal-regulated protein kinase
In the present study, Real Time quantitative PCR (RT-qPCR) was performed to study the fold change analysis for evaluating differential expression of genes. In this, gene expression normalization was done by using GAPDH constitutive genes by determining delta Ct (cycle threshold). In a RTPCR assay, a positive reaction is detected by the accumulation of a fluorescent signal, and Ct is defined as the number of cycles required for the fluorescent signal to cross the threshold (i.e., exceeds background level). Ct levels are inversely proportional to the amount of target nucleic acid in the sample, i.e., lower the value of Ct, higher the amount of target nucleic acid in the sample.
Then, delta Ct was evaluated which is the difference of average Ct of target gene (TIMP1 and ERK gene) and constitutive gene (GAPDH gene):
ΔCt = Average Ct_Target-average Ct_Normalizer.
Again, the difference of mean Ct values of control and cases was determined, which is delta delta Ct: ΔΔCt = ΔCt_case−ΔCt control
After this, true FC was represented to compare the expression of genes between cases and controls by the following:
FC = 2−ΔΔCt
If FC >1, true fold change = FC and
If FC <1, true fold change = −1/FC.
Sample size calculation
The sample size could not be calculated as it is a pilot study. Sample size was determined based upon the number of patients either reporting to pain clinic or referred to pain clinic. Since the study was limited to 18 months and considering at least 10–12 patients per month, a minimum of 200 patients could be included.
Statistical analysis
Statistical analysis was done using the SPSS version 20 [Statistical Product and Service Solutions, owned by International Business Machines Corporation (IBM) HQ: Armonk New York US. Currently officially “IBM® SPSS® Statistics”]. Delta Ct value is inversely proportional to the nucleic acid content, so on the basis of Δct value for gene expression study, depending on normality, unpaired students t-test (for normally distributed variables) and Mann–Whitney U-test was used to compare NP from NNP. For comparing statistical significance of expressional analysis, unpaired t-test was used to compare Δct of NP and NNP group.
RESULTS
A total of 285 patients with chronic pain were assessed. Out of which, 153 patients were noted to be having NP and 132 had NNP. The various clinical diagnoses of both the groups are depicted in Figures 1 and 2, and the demographic characteristics of the patients are shown in Table 3.
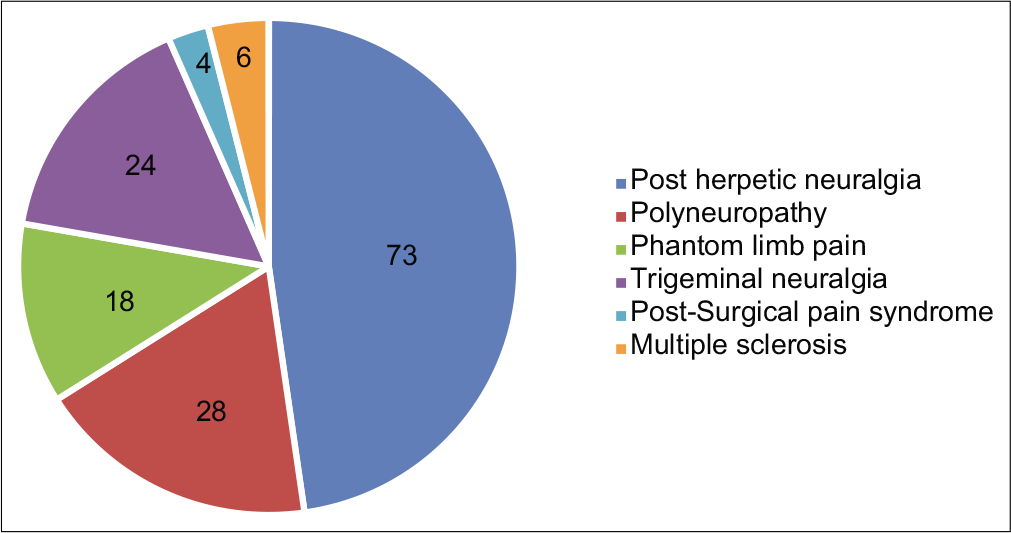
- Various clinical diagnosis in “Neuropathic pain” group.
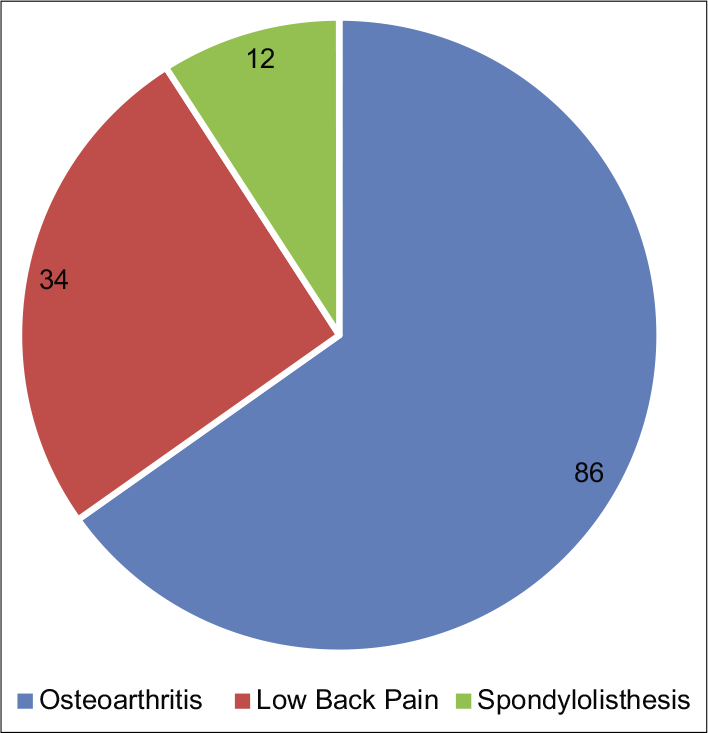
- Various clinical diagnosis in “nonneuropathic pain” group.
| Demographic variables | Total (n=285) | NP (n=153) | NNP (n=132) |
|---|---|---|---|
| Age (in years) | 51.10±12.21 | 54.10±12.18 | 48±10.34 |
| Gender (females) (%) | 157 (55) | 71 (46) | 86 (65) |
| Duration of pain (in months) | 11.53 | 15.79 | 10.32 |
| Baseline avg. VAS score of pain | 6.0 | 6.7 | 5.6 |
VAS: Visual analogue scale, NP: Neuropathic, NNP: Nonneuropathic pain
The average duration of chronic pain was 11 months for 285 patients. Duration of pain was higher in patients with NP. Majority of the patients with the diagnosis of NP had VAS score higher than that of patients with NNP. The mean VAS score of all the patients was 6.0 [Table 3].
A baseline venous sample (2 ml) was withdrawn from each patient including both NP and NNP at the time of presentation. The gene expression analysis was done using qRT-PCR.
The Delta Ct values for ERK and TIMP1 are expressed in mean ± standard deviation in Table 4. The mean ΔCt of ERK was significantly down regulated reflecting the higher nucleic acid content in the target gene in patients with NP. On the contrary, the mean ΔCt of TIMP1 gene was significantly upregulated in patients with NP when compared to NNP reflecting the lower nucleic acid content in the target gene.
| Gene under evaluation | Group NP | Group NNP | Level of significance P* |
|---|---|---|---|
| Δct ERK | 3.11±2.03 | 4.13±1.80 | <0.0001 |
| Δct TIMP1 | 4.81±1.79 | 3.41±1.77 | <0.0001 |
NP: Neuropathic, NNP: Nonneuropathic pain, SD: Standard deviation
Further, the ΔΔCt values and the respective FCs were calculated [Table 5]. The mRNA expression of TIMP1 gene is significantly down regulated (2.65-fold) (P ≤ 0.01) in NP patients when compared with NNP. The mRNA expression level of ERK is significantly up-regulated (2.03-fold) (P ≤ 0.01) when compared with NNP group. Both the aforementioned changes were found to be statistically significant (P < 0.05). The amplification with the corresponding cycle is depicted in Figure 3, whereas Figure 4 depicts the melt peak curve for both the ERK and TIMP1 gene against temperature.
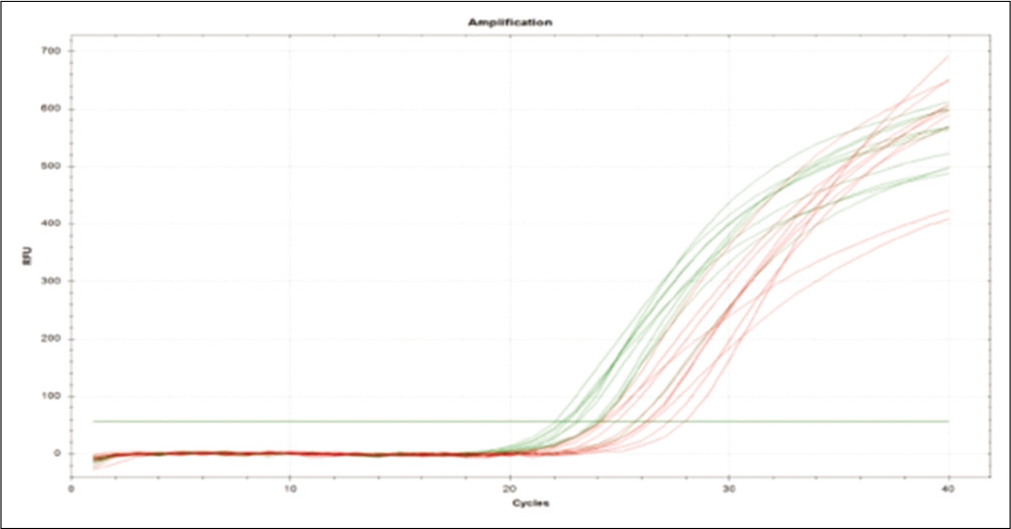
- Amplification plot of extracellular signal-regulated protein kinase and tissue inhibitors of matrix metalloproteases 1 gene.
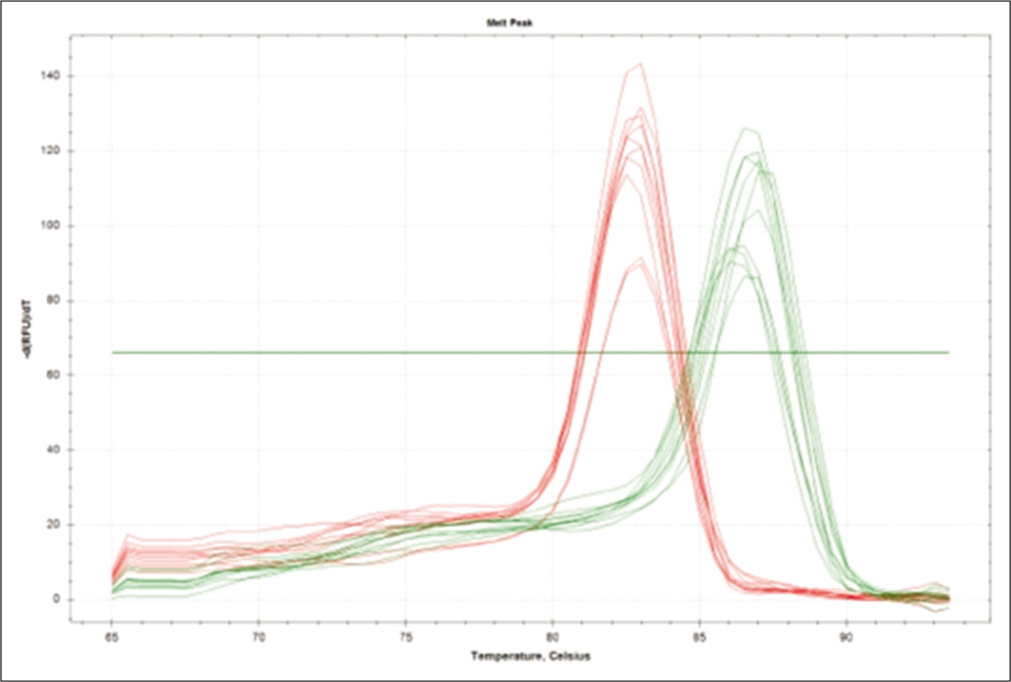
- Melt peak curve of extracellular signal-regulated protein kinase and tissue inhibitors of matrix metalloproteases 1 gene.
| ΔΔCt ERK | ΔΔCt TIMP1 | Fold change ERK | Fold change TIMP1 |
|---|---|---|---|
| −1.02 | 1.4 | ↑2.03 (p=0.01)* | ↑2.65 (p=0.01) * |
DISCUSSION
The present study is the first human study to evaluate the modulation of ERK and TIMP1 gene modulation in the pathogenesis of NP. The results of the present study showed that mRNA expression of TIMP1 gene was significantly (P < 0.05) down regulated in NP patients, whereas mRNA expression of ERK gene was significantly up regulated in patients with NP when compared to NNP.
An upcoming area in pain research is to find the predisposing factors and the genes responsible for NP by observing the sensitivity of various nociceptive stimuli among individuals. The genetic testing would allow early identification of patients at risk, and thus, help in the development of prophylactic and therapeutic intervention.
In a study by Fujimoto et al., TIMP-1 was shown to inhibit MMP-9 activity, suggesting its induction may have a neuroprotective function in CNS insults.[20] Interestingly, elevated levels of TIMP expression, induced through activation of cannabinoid receptors, was shown to reduce cancer cell invasiveness. This suggests that cannabinoids may provide a therapeutic approach to the attenuation of NP development.[21] Both these study findings corroborate with that of our study where there was a negative upregulation of TIMP1 as depicted by the flow change in patients with chronic pain when NP was compared to NNP.
In the study by Ji et al., on intracellular kinases, it was observed that nociceptor stimulus led to membrane receptor activation of kinases including protein kinases A, C and mitogen-activated protein kinases (MAPKs) leading to critical membrane phosphorylation and alteration of neuronal activity. MAPKs are important for intracellular signal transduction and play critical roles in regulating neural plasticity and inflammatory responses. MAPK family consists of three major members, extracellular signal regulated kinases ERK, p38, and JNK kinase. MAPK activation can lead to alteration in the gene expression and persistent neuronal function changes that underlie chronic pain associated sensitization. Animal studies have suggested that activation of ERK pathways contributes to NP.[22-24]
ERK is one of the three groups of MAPK system and is activated by neuronal activity. The balance between the expression of MMP and their endogenous tissue inhibitors (TIMP1) has important implication for the healthy nervous system as well as pathogenesis and recovery in disease or injury. MMP expression and activity is controlled by intracellular and extracellular signaling molecules, cell surface receptor activity, transcription factor proteolytic activation of inactive MMP proform and inhibition by TIMPs. There is evidence that MMP and TIMP1 play a significant role in chronic conditions such as NP.[25,26]
Moreover, the present study happens to be the very first study ever undertaken on human beings for the evaluation of modulation of mRNA expressions of TIMP1 in chronic NP. The sample for gene expression was evaluated as the baseline in all the patients and comparison was done between patients with NP and NNP. It was observed that the mRNA expression of TIMP1 gene was significantly down regulated in NP patients (FC = 2.03 and P = 0.01) and mRNA expression of ERK gene was significantly up-regulated (FC = 2.65 and P = 0.01) in patients with NP when compared to NNP values.
Our finding for expression of ERK is in concordance to Zhang et al., who clearly demonstrated that expression of ERK are upregulated in the dorsal root of spinal cord of rats after peripheral axonotomy. Similarly, a significant upregulation in ERK gene expression was observed by Saxena et al. in human participants of chronic persistent postsurgical pain following staging laparotomy for carcinoma ovary.[19,27]
Limitation
First, the decision of the diagnosis of NP was done by only one expert, wherein, the decision of two experts could have been considered for the diagnosis. Second, the data was single centric.
CONCLUSION
The results of the present study suggest that the mRNA expression of TIMP1 gene was significantly (P < 0.05) down regulated in NP patients, whereas mRNA expression of ERK gene was significantly up regulated in patients with NP when compared to NNP indicating its potential role in the modulation and pathogenesis of NP. However, we suggest a prospective multi-centric trial with a larger sample size to confirm the findings of the present study.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- The genetics of neuropathic pain from model organisms to clinical application. Neuron. 2019;104:637-53.
- [CrossRef] [Google Scholar]
- prevalence of chronic pain with or without neuropathic characteristics in the general population. Pain. 2008;136:380-87.
- [CrossRef] [Google Scholar]
- Prevalence of self-reported neuropathic pain and impact on quality of life: A prospective representative survey. Acta Anaesthesiol Scand. 2008;52:132-6.
- [CrossRef] [Google Scholar]
- Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain. 2014;155:654-62.
- [CrossRef] [Google Scholar]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789-858.
- [CrossRef] [Google Scholar]
- Improving the translation of analgesic drugs to the clinic: Animal models of neuropathic pain. Br J Pharmacol. 2014;171:2951-63.
- [CrossRef] [Google Scholar]
- Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128:1847-54.
- [CrossRef] [Google Scholar]
- The effect of SCN9A variation on basal pain sensitivity in the general population: An experimental study in young women. J Pain. 2015;16:971-80.
- [CrossRef] [Google Scholar]
- A novel SCN9A mutation responsible for primary erythromelalgia and is resistant to the treatment of sodium channel blockers. PLoS One. 2013;8:e55212.
- [Google Scholar]
- Neuropathic pain as part of chronic widespread pain: Environmental and genetic influences. Pain. 2015;156:2100-6.
- [CrossRef] [Google Scholar]
- Neuropathic pain: Role of inflammation, immune response, and ion channel activity in central injury mechanisms. Ann Neurosci. 2012;19:125-32.
- [CrossRef] [Google Scholar]
- Brain regional and cellular localization of gelatinase activity in rat that have undergone transient middle cerebral artery occlusion. Neuroscience. 2008;152:8-17.
- [CrossRef] [Google Scholar]
- Changes in the expression of extracellular matrix (ECM) and matrix metalloproteinases (MMP) of proliferating rat parotid acinar cells. J Dent Res. 1998;77:1504-14.
- [CrossRef] [Google Scholar]
- Regulation and function of matrix metalloproteinases in nervous system injury and neuropathic pain. Ann Neurosci. 2010;15:94-105.
- [CrossRef] [Google Scholar]
- Matrix metalloproteinases and their inhibitors in human traumatic spinal cord injury. BMC Neurol. 2007;7:17.
- [CrossRef] [Google Scholar]
- The ERK/MAPK pathway, as a target for the treatment of neuropathic pain. Expert Opin Ther Targets. 2005;9:699-713.
- [CrossRef] [Google Scholar]
- Chronic persistent post-surgical pain following staging laparotomy for carcinoma of ovary and its relationship to signal transduction genes. Korean J Pain. 2016;29:239-48.
- [CrossRef] [Google Scholar]
- Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J Cerebral Blood Flow Metabolism. 2008;28:1674-85.
- [CrossRef] [Google Scholar]
- Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J Natl Cancer Inst. 2008;100:59-69.
- [CrossRef] [Google Scholar]
- Neuronal plasticity and signal transduction in nociceptive neurons: Implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1-10.
- [CrossRef] [Google Scholar]
- Identification of DGAT2 inhibitors using mass spectrometry. J Biomol Screen. 2016;21:117-26.
- [CrossRef] [Google Scholar]
- Molecular detection and characterization of zoonotic and veterinary pathogens in ticks from North Eastern China. Front Microbiol. 2016;7:1913.
- [CrossRef] [Google Scholar]
- Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol Sci. 2009;30:336-40.
- [CrossRef] [Google Scholar]
- Regulation and function of matrix metalloproteinases in nervous system injury and neuropathic pain. Ann Neurosci. 2008;15:534-42.
- [CrossRef] [Google Scholar]
- Gene array analysis to determine the components of neuropathic pain signaling. Curr Opin Mol Ther. 2005;7
- [Google Scholar]




